6. Optional: Modelling a novel ligand
If the ligand is new or it does not exist in the CCP4 Monomer library, then it is necessary to create a description for the new ligand. This is done using the program AceDRG. For the purpose of the tutorial let’s pretend that PTQ is a new ligand, and we give it a generic name DRG.
Select the AceDRG job from the Create Ligand category and enter the following options:
SMILES String:: c1ccc(cc1)CCS[C@H]2[C@@H]([C@H]([C@H]([C@H](O2)CO)O)O)O
Monomer code:: DRG
NOTE: This is an arbitrary name for the ligand. Some software will complain if you use a code that already exists in the monomer library. The wwPDB has reserved some codes (DRG, INH, LIG and 01 – 99) that can safely be used for new ligands.
Press Run to create the ligand restraints file. We must now download the ligand description to the local machine. Go to the job’s I/O tab.
AceDRG generates three outputs: the stereochemical restraints for the new ligand (DRG_acedrg.cif) and atomic coordinates for an example low-energy conformation in both mmCIF and PDB formats (DRG_acedrg.mmcif and DRG_acedrg.pdb) that can be used as a starting point for modelling the ligand.
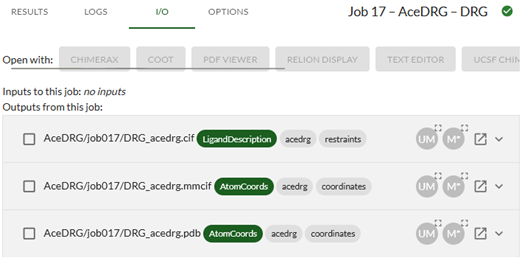
Click on the Open in new window icon
for AceDRG/jobXXX/DRG_acedrg.cif. This will open the CIF file in your browser. You can save the file to your local machine (File-->Save as).
When running Moorhen, you can insert the ligand into the map with Ligand -> Import dictionary...
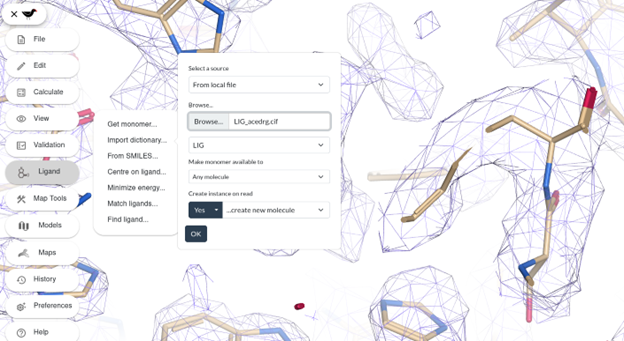
You can place the ligand into the density then fit, refine and merge the ligand into the model as described previously.
If the ligand is not in the monomer library, when running Servalcat with a new ligand included in your model, you will need to provide the corresponding stereochemical restraints:
CIF dictionaries:: AceDRG/jobXXX/DRG_acedrg.cif
Note that the atom names in the ligand are important for the stereochemical restraints to be applied correctly during refinement. If you are working with known ligands, this will usually not be a problem, but if you create new restraints (for example as we’ve just done with AceDRG), the atom names might not match correctly with the “same” ligand molecule in any other structures. To avoid errors, make sure you don’t mix and match your ligand information from different sources.